
Crianças Especiais: Pequenos Anjos Disfarçados entre
nós!
"Como Nascem os Anjos" é uma história inspirada na vida de uma criança especial cujo coração não conhece o significado da palavra desistir e que distribui por onde passa sorrisos mágicos.
"Como Nascem os Anjos" é uma história inspirada na vida de uma criança especial cujo coração não conhece o significado da palavra desistir e que distribui por onde passa sorrisos mágicos.
As passagens relatadas nos levam a questionar quais segredos são guardados pelas vidas de crianças
especiais.
Seriam somente o que aparentam fisicamente ou estariam disfarçados a serviço da GRAÇA, no propósito de
nos ensinar inúmeras lições?
O caráter de uma doença é o resultado da ação da combinação de
fatores genéticos e ambientais, porém é conveniente distinguir se a causa
principal é uma transformação genética, ou uma combinação de pequenas variações
que, juntas podem produzir um defeito sério.
A Genética atual tem visto o seu campo de ação ampliado: cerca
de 30% das admissões em Pediatria e cerca de 10% das admissões de adultos em
hospitais. Isto deve-se a vários fatores, principalmente à melhoria das
condições de saúde que, favorecendo a sobrevida de indivíduos com algumas
síndromes, tornam visíveis algumas doenças genéticas.
A incidência de indivíduos com síndromes genéticas é alta: 14%
dos recém-nascidos tem um defeito congênito leve único, não valorizável, e 0,8
% tem dois defeitos. O aumento da incidência relativa das anomalias de ordem
genética têm algumas causas: existe hoje maior acesso e capacidade de
diagnóstico (mais especialistas e mais meios); a seleção natural deixa de ser
operativa com crianças doentes que antes morriam cedo e atingem hoje a idade da
procriação.
Durante vários anos foram feitas pesquisas sobre as doenças
genéticas e foram identificadas mais de 50 tipos de doenças e aberrações cromossômicas dentre as quais destacamos algumas:

É
uma doença genética extremamente rara que acelera o processo de envelhecimento
em cerca de sete vezes em relação à taxa normal.
A expectativa média de vida das
pessoas é de 14 anos para as meninas e 16 para os meninos.
Essa doença afeta 1 entre 8 milhões de crianças e
desde a sua identificação foram relatados cerca de 100 casos.
Características
fenotípicas
Os recém-nascidos apresentam o choro fraco,
semelhante ao miado do gato, por isso a denominação da Síndrome Cri du Chat.
- Também apresentam baixo peso ao nascimento devido a um retardo de crescimento intra-uterino;
- Microcefalia Face arredondada;
- Hipertelorismo (aumento da distância entre os olhos);
- Prega epicântica (presente no canto interno dos olhos);
- Estrabismo;
- Micrognatia (desenvolvimento reduzido da mandíbula);
- Orelhas de baixa implantação (abaixo da linha do nariz);
- Malformações dentárias (geralmente os dentes são projetados para frente devido a alterações no desenvolvimento crânio-facial);
- Luxação de quadril;
- Prega palmar transversal única;
- Podem ocorrer problemas cardíacos e/ ou renais;
- Pé torto congênito;
- Hipotonia (tônus muscular diminuído), na maioria dos casos;
- Apresenta freqüentes dificuldades na alimentação, como pouca força para sugar, freqüente engasgo e refluxo.
- São bastante suscetíveis a infecções respiratórias e gastrintestinais;
- Hipodesenvolvimento pondero-estatural (peso e estatura reduzidos).
- Déficit cognitivo;
- Dificuldade de concentração/atenção,
- agitação e irritabilidade, sono agitado.
- Atraso no desenvolvimento da aquisição da linguagem.
É fundamental deixar claro que nem todos os
portadores de Cri du Chat terão todas essas características, elas dependerão da
quantidade de estímulos motores e cognitivos que receberem e da quantidade de
perda do material genético do braço curto do cromossomo.
Síndrome Klinefelter

Uma em cerca de mil crianças nasce com um cromossomo X extra
(47, XXY), que caracteriza a síndrome de Klinefelter,
resultante, geralmente, de uma não-disjunção na formação do óvulo. Embora
apresente cromatina sexual, é do sexo masculino (este é determinado pelo
cromossomo Y, mesmo quando acompanhado de mais de um cromossomo X). Mas a
fertilidade é baixa, com nenhuma ou pouca produção de espermatozóides (os
testículos são pouco desenvolvidos) e, às vezes, desenvolvimento exagerado das
glândulas mamárias (ginecomastia). A altura é acima da média. O tratamento
hormonal pode ajudar a diminuir esses sintomas, mas não a baixa fertilidade.
Características:
Embora possam ter ereção e ejaculação, são estéreis, pois seus
testículos são pequenos e não produzem espermatozóides devido à atrofia dos
canais seminíferos. Outras características muitas vezes presentes são: estatura
elevada corpo eunucóide, pênis pequeno, pouca pilosidade no púbis e
ginecomastia (crescimento das mamas). Além dessas alterações do sexo
fenotípico, os pacientes com Síndrome de Klinefelter apresentam uma evidente
diminuição do nível intelectual, sendo esta tanto mais profunda quanto maior
for o grau da polissomia. Ao contrário do que ocorre na Síndrome de Tuner, os
pacientes klinefelter apresentam problemas no desenvolvimento da personalidade,
que é imatura e dependente, provavelmente em decorrência de sua inteligência
verbal diminuída. As dificuldades de relacionamento interpessoal incluem, por
vezes, alterações no processo de identificação psicossexual, envolvendo casos
de homossexualismo e transexualismo. Fisicamente são quase indistinguíveis dos
homens com cariótipo 46,XX.
Síndrome de Turner

A síndrome de Turner é uma patologia cromossômica caracterizada
por um fenótipo feminino, com baixa estatura, infantilismo sexual e certo
aspecto intelectual questionável. As causas genéticas são, em termos numéricos,
os determinantes mais importantes das malformações congênitas, embora problemas
durante o nascimento e anomalias congênitas também sejam expressões comumente
empregadas para descrever problemas durante o desenvolvimento. A cada 2.500
nascimentos uma criança apresenta o fenótipo característico da síndrome de
Turner atingindo 1.500.000 mulheres no mundo.
A síndrome de Turner caracteriza-se como uma anormalidade
genética expressa através de uma monossomia (presença de apenas um cromossomo
sexual) cujas portadoras apresentam fenótipo feminino e características
fenotípicas peculiares. O estudo do cariótipo das portadoras desta síndrome
normalmente indica 45 cromossomos, onde há apenas um X no que deveria ser um
par de cromossomos sexuais (XX). Foi avaliado que os fatores genéticos causam
aproximadamente 1/3 de todas as irregularidades no nascimento e quase 85% das
anomalias com causa conhecida. No máximo 1% dos embriões femininos com
monossomia do X se mantêm vivos. A síndrome de Turner causa 18% dos abortos
espontâneos cromossomicamente anormais, sendo esta monossomia do cromossomo
sexual de origem materna, o que indica o erro meiótico como paterno. A falha na
gametogênese (não-disjunção) que causa a monossomia do X, quando é detectada, está
no gameta do pai, em 75% dos casos.
As portadoras da síndrome de Turner freqüentemente apresentam
atraso na primeira menstruação, além de déficits no desenvolvimento de outras
características marcantes associadas à puberdade devido a ausência de hormônios
importantes para um desenvolvimento sexual normal. Dentre os sintomas físicos
mais comuns encontramos a presença de pescoço alado e a baixa estatura como
características marcantes. O gene responsável pelo crescimento dos ossos se
encontra ausente nas paciente de síndrome de Turner. A maior estatura relatada
foi de uma mulher com 1,40 m de altura. Se hormônios forem administrados antes
da puberdade (em torno dos 8 anos de idade), algumas portadoras podem atingir
uma estatura mais alta, embora essa estatura varie bastante entre as
portadoras. Outro sintoma característico é o pescoço alado, tipicamente menor
que o normal e apresentando pele com uma textura que confere um aspecto “alado”
à aparência da portadora. Somado a esses sintomas existem outras
características físicas incluídas na síndrome: disgenesia gonadal, face incomum
típica, linha posterior de implantação dos cabelos baixa, tórax largo com
mamilos amplamente espaçados. Algumas podem apresentar anormalidades cardíacas
e renais, pressão sanguínea alta, obesidade, diabetes mellitus , catarata,
problemas na glândula tireóide e artrite.
O ciclo menstrual também é afetado na síndrome de Turner. O
ciclo pode ser atrasado até a alta adolescência ou começar normalmente e então
sutilmente ir diminuindo. Embora a maior parte das mulheres com a síndrome de
Turner seja infértil, a gravidez tem sido raramente relatada. Como a puberdade
é freqüentemente atrasada ou comprometida, as características sexuais
secundárias (desenvolvimento das mamas) são diminuídas ou limitadas. A
apresentação de um quadro clínico difere com a idade e pode ser dividido em
quatro estágios: período neonatal, infância, adolescência e vida adulta. O
recém-nascido apresenta linfedema, má-formação cárdio-vascular e face
característica. Na infância, o achado para o diagnóstico é a baixa estatura. Na
adolescência e na idade adulta, as queixas mais comuns estão relacionadas à insuficiência
gonadal, sendo sempre o diagnóstico diferencial realizado através do cariótipo.
Entretanto, existem casos em que as características detectadas pelo ultra-som
durante o pré-natal podem ser relacionadas a outras anomalias genéticas. Para
esses casos a suspeita de uma possível síndrome de Turner poderá ser confirmada
com a coleta do líquido amniótico. Já em crianças, atesta-se pelo cariótipo de
cada uma. No caso de o cariótipo no sangue de uma menina portadora da síndrome
de Turner, estar normal é preciso que se analise outro tecido. Desta forma, a
síndrome de Turner poderá ser identificada ainda pelo cariótipo em fibroblastos
e tecido epidermal, obtidos por biópsia.
Personalidade das
Portadoras da Síndrome
É observado que a maior parte das meninas portadoras da síndrome
de Turner apresentam desempenho mediano ou até mesmo acima da média escolar, o
que ocorre, em geral, pela dedicação que desenvolvem para com a escola.
Entretanto, algumas apresentam dificuldades em geometria e aritmética, tal como
em entender um mapa ou desenhar uma figura; ocasionado principalmente por
déficit de percepção espacial e visual-motor. A maioria das portadoras
desenvolve inteligência compatível com pessoas da mesma idade, sendo encontrado
em apenas uma pequena parcela delas um leve retardamento mental. Por esse
motivo, antigamente a síndrome era associada ao retardo mental. Somente com
estudos mais atuais essa idéia foi desmistificada. Deste modo, não existe
nenhum déficit intelectual em pacientes da síndrome de Turner, embora
apresentem melhor performance na área verbal, em comparação com o desempenho
perceptivo e espacial, que pode ser deficitário.
O relacionamento inter-pessoal com professores e colegas varia
de bom a muito bom. Entretanto, periodicamente podem ser importunadas a
respeito de sua baixa estatura. Por outro lado, a maturidade emocional das
portadoras da síndrome de Turner apresenta-se tardiamente, principalmente se
comparada com o desenvolvimento emocional observado em suas irmãs e amigas.
No Brasil, os hospitais públicos que fornecem tratamento para
essa anomalia genética, consideram a síndrome de Turner menos grave do que
outras alterações cromossômicas. Sendo assim, os pais que têm uma filha
portadora ao buscar um tratamento hormonal para elas, encontram grandes
dificuldades, como a fila de espera para obter o tratamento de reposição
hormonal necessário para aumentar sua estatura e desenvolver suas
características sexuais secundárias. Neste contexto seria importante ser
iniciado um acompanhamento psicológico para a portadora da síndrome de Turner,
bem como para sua família. Outro ponto importante é a inserção da portadora e
de sua família em um grupo de contato, pois já é conhecido o progresso
psico-social que este procedimento proporciona nos desempenhos escolares,
sociais e familiares, além de elevar a auto-estima que pode estar debilitada.
SÍNDROME DE "X" FRÁGIL

É causada mais frequentemente por comprometimento
mental com caráter hereditário, afetando o desenvolvimento intelectual e o
comportamento em geral de homens e mulheres.
A expressão "x" frágil deve-se a uma
anomalia causada por um gene defeituoso localizada no cromossomo X, que por sua
vez, passa a apresentar uma falha numa de suas partes. O X está presente no par
de cromossomos que determina o sexo (xy nos homens e xx nas mulheres).
Essa falha ou "fragilidade" do
"X" causa um conjunto de sinais e sintomas clínicos (ou uma
síndrome). Daí o nome de síndrome do X frágil (sxf).
Os meninos e as meninas podem herdar um X frágil
(normalmente de uma mãe portadora), mas os meninos, sem a influência da
potencialidade dominante de um X normal, são muitos suscetíveis às
conseqüências intelectuais ou comportamentais negativas.
A criança afetada parece correr um risco
consideravelmente aumentado de retardo mental; as estimativas atuais são de
que, entre os homens, 5 a 7 por cento de todos os retardos são causados por
esta síndrome (Zigler e Hodapp, 1991).
SÍNDROME DE ANGELMAN

Essa anomalia ocorre no espaço entre 11 e 13 do
braço "q" do cromossomo 15 ou quando ganha 2 cromossomos do pai.
As crianças que apresentam esse tipo de anomalia
tem alguns sintomas como: andar desajeitado, risadas freqüentes, convulsões,
perímetro cefálico pequeno e achatamento occipital.
Não há prevalência de sexo entre os afetados.
O diagnóstico clinico precoce é raro e difícil. O
quadro clínico evolutivo vai se evidenciando com o passar do tempo, tornando o
diagnóstico mais fácil com a evolução da síndrome.
A síndrome é caracterizada por: atraso no
crescimento, a criança apresenta peculiar fragilidade com freqüência, em
brancos ela costuma apresentar hipogmentação na pele, nos olhos e nos
cabelos.
SÍNDROME DE APERT

É um defeito genético e faz parte das quase 6.000
síndromes genéticas conhecidas. Pode ser herdada de um dos pais ou por mutação
nova.
Ocorre em aproximadamente 1 para 160.000 a 200.000
nascidos vivos. Sua causa se encontra em uma mutação durante o período de
gestação, nos fatores de crescimento dos fibroblastos que ocorre durante o
processo de formação dos gametas.
O crânio tem fusão prematura e é incapaz de
desenvolver-se normalmente, o terço médio da face (área que vai da órbita do
olho até o maxilar superior) parece retraída ou afundada, os dedos das mãos e
dos pés tem vários graus. Essa síndrome foi classificada como anomalia
craniofacial.
Síndrome de Down


A síndrome de Down é
um distúrbio genético que ocorre ao acaso durante a divisão celular do
embrião. Esse distúrbio ocorre, em média, em 1 a cada 800 nascimentos e
tem maiores chances de ocorrer em mães que engravidam quando mais
velhas. É uma síndrome que atinge todas as etnias.
Em uma célula normal da espécie humana existem 46 cromossomos divididos
em 23 pares. A pessoa que tem síndrome de Down possui 47 cromossomos,
sendo que o cromossomo extra é ligado ao par 21. Esse distúrbio
genético pode se apresentar das seguintes formas:
Trissomia 21 padrão
Cariótipo 47XX ou 47XY (+21). O indivíduo apresenta 47 cromossomos em
todas as suas células, tendo no par 21 três cromossomos. Ocorre
aproximadamente em 95% dos casos.
Trissomia por translocação
Cariótipo 46XX (t14;21) ou 46XY (t14; 21). O indivíduo apresenta 46
cromossomos e o cromossomo 21 extra se adere a outro par, geralmente o
14. Ocorre aproximadamente em 3% dos casos.
Mosaico
Cariótipo 46XX/47XX ou 46XY/47XY (+21). O indivíduo apresenta células
normais (46 cromossomos) e células trissômicas (47 cromossomos). Ocorre
aproximadamente em 2% dos casos.
Essa síndrome é diagnosticada logo após o nascimento, pelo médico
pediatra que analisa as características fenotípicas comuns à síndrome.
A confirmação da síndrome é dada por meio de uma análise citogenética.
Não existem graus da síndrome de Down, porém o ambiente familiar, a
educação e a cultura em que a criança está inserida influenciam muito
no seu desenvolvimento.
Alguns exames feitos pela gestante no pré-natal também podem
identificar se o bebê será ou não portador desse distúrbio genético.
Alguns exames que podem ser feitos são:
- Amostra de Vilocorial: consiste em uma amostra do tecido placentário obtido via vaginal ou via abdome. Nesse exame há risco de aborto.
- Amniocentese: consiste na coleta de líquido amniótico que será observado em uma análise cromossômica. O líquido amniótico é retirado por aspiração com uma agulha inserida na parede abdominal até o útero. Nesse exame, o risco de aborto é menor.
- Dosagem de alfafetoproteína materna: observou-se que o nível baixo de alfafetoproteína no sangue materno indica desordens cromossômicas, em particular a síndrome de Down. Se o nível baixo for detectado, outros exames deverão ser realizados.
Os cuidados com uma criança que possui a síndrome de Down não se
diferenciam em nada com os cuidados destinados a crianças que não
possuem essa síndrome. Especialistas recomendam aos pais que estimulem
a criança a ser independente, conforme cresce. Ela deve ser tratada com
naturalidade, respeito e carinho como qualquer criança.
SÍNDROME DE WILLIAMS

é uma desordem
genética que, talvez, por ser rara, freqüentemente não é diagnosticada. Sua
transmissão não é genética. O nome desta síndrome vem do médico, Dr. J.C.P.
Williams que a descreveu em 1961 na Nova Zelândia e pelo Dr. A. J. Beuren da
Alemanha em 1962 .
Acometendo ambos os sexos, na maioria dos casos infantis (primeiro ano de
vida), as crianças têm dificuldade de se alimentar, ficam irritadas facilmente
e choram muito.
A síndrome de Williams é uma doença caracterizada por "face de gnomo ou fadinha”, nariz pequeno e empinado, cabelos encaracolados, lábios cheios, dentes pequenos e sorriso freqüente. Estas crianças normalmente têm problemas de coordenação e equilíbrio, apresentando um atraso psicomotor. Seu comportamento é sociável e comunicativo embora utilizem expressões faciais, contatos visuais e gestos em sua comunicação.
Embora comecem a falar tarde, por volta dos 18 meses, demonstram facilidade para aprender rimas e canções, demonstrando muita sensibilidade musical e concomitantemente boa memória auditiva. Seu desenvolvimento motor é mais lento. Demoram a andar, e tem grande dificuldade em executar tarefas que necessitem de coordenação motora tais como: cortar papel, desenhar, andar de bicicleta, amarrar o sapato etc..
Tratamento e Prevenção das Complicações
É muito importante identificar portadores desta síndrome logo na primeira infância, pois, tem influência em diversas partes do desenvolvimento cognitivo, comportamental e motor.
As medidas preventivas devem-se iniciar logo após o diagnóstico com um estudo minucioso para descarte de anomalias do coração e rins. É necessário monitorar freqüentemente a hipertensão arterial, incluindo a avaliação da tensão arterial nos quatro membros.
A otite crônica exige avaliações auditivas freqüentes e quando necessário o envio para uma consulta de otorrinolaringologia. O tratamento de problemas dentários necessita da profilaxia da endocardite. Face às infecções urinárias freqüentes torna-se necessário avaliar a função renal periodicamente e realizar um estudo minucioso na infância e na adolescência. Na adolescência, para além de se manter a vigilância dos sistemas já descritos, deve-se pesquisar a presença de escoliose e contratura das articulações. Os problemas alimentares observados nos mais novos são ultrapassados, sendo a obesidade encontrada em 29% dos adultos. O comportamento e aproveitamento escolar, quando problemáticos carecem de medidas de apoio. A ansiedade pode estar associada à úlcera péptica e a litíase biliar é um diagnóstico possível em doentes com dores abdominais.
A síndrome de Williams é uma doença caracterizada por "face de gnomo ou fadinha”, nariz pequeno e empinado, cabelos encaracolados, lábios cheios, dentes pequenos e sorriso freqüente. Estas crianças normalmente têm problemas de coordenação e equilíbrio, apresentando um atraso psicomotor. Seu comportamento é sociável e comunicativo embora utilizem expressões faciais, contatos visuais e gestos em sua comunicação.
Embora comecem a falar tarde, por volta dos 18 meses, demonstram facilidade para aprender rimas e canções, demonstrando muita sensibilidade musical e concomitantemente boa memória auditiva. Seu desenvolvimento motor é mais lento. Demoram a andar, e tem grande dificuldade em executar tarefas que necessitem de coordenação motora tais como: cortar papel, desenhar, andar de bicicleta, amarrar o sapato etc..
Tratamento e Prevenção das Complicações
É muito importante identificar portadores desta síndrome logo na primeira infância, pois, tem influência em diversas partes do desenvolvimento cognitivo, comportamental e motor.
As medidas preventivas devem-se iniciar logo após o diagnóstico com um estudo minucioso para descarte de anomalias do coração e rins. É necessário monitorar freqüentemente a hipertensão arterial, incluindo a avaliação da tensão arterial nos quatro membros.
A otite crônica exige avaliações auditivas freqüentes e quando necessário o envio para uma consulta de otorrinolaringologia. O tratamento de problemas dentários necessita da profilaxia da endocardite. Face às infecções urinárias freqüentes torna-se necessário avaliar a função renal periodicamente e realizar um estudo minucioso na infância e na adolescência. Na adolescência, para além de se manter a vigilância dos sistemas já descritos, deve-se pesquisar a presença de escoliose e contratura das articulações. Os problemas alimentares observados nos mais novos são ultrapassados, sendo a obesidade encontrada em 29% dos adultos. O comportamento e aproveitamento escolar, quando problemáticos carecem de medidas de apoio. A ansiedade pode estar associada à úlcera péptica e a litíase biliar é um diagnóstico possível em doentes com dores abdominais.
Personalidade e comportamento
Nas crianças portadoras desta síndrome é grande a sociabilidade,
entusiasmo, grande sensibilidade, tem uma memória fantástica para pessoas,
nomes e local; ansiedade medo de alturas, preocupação excessiva com
determinados assuntos ou objetos, distúrbios do sono, controle do esfíncter É
normal crianças com esta síndrome serem amigas de adultos e procurarem a
companhia deles ao mesmo tempo tem dificuldade em fazer amizades outras
crianças da sua idade. Muitas crianças com esta síndrome demonstram medo ao
escutarem ruídos de bater palmas, liquidificador, avião, etc., por serem
hipersensíveis ao som.
LIBRAS

“AS MÃOS ROMPEM O SILÊNCIO E FAZEM A COMUNICAÇÃO DE QUEM NÃO OUVE, mas vê, sente e se emociona”
PSICOMOTRICIDADE


Livro da minha professora e coordenadora do Curso
Afasia
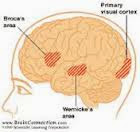
A afasia é por si só a perda da capacidade e das habilidades de linguagem falada e escrita. É um sintoma comum na neurologia clínica, muitas vezes como conseqüência de um acidente vascular cerebral cuja localização geralmente se dá junto à artéria cerebral média esquerda ou nos ramos junto à região do cérebro responsável pela linguagem
Dislalia

A dislalia é um distúrbio da fala, caracterizado pela dificuldade em articular as palavras. Basicamente consiste na má pronúncia das palavras, seja omitindo ou acrescentando fonemas, trocando um fonema por outro ou ainda distorcendo-os ordenadamente.A falha na emissão das palavras pode ainda ocorrer em fonemas ou sílabas. Assim sendo, os sintomas da Dislalia consistem em omissão, substituição ou deformação dos fonemas.
Dislexia

Dislexia é uma específica dificuldade de aprendizado da Linguagem: em Leitura, Soletração, Escrita, em Linguagem Expressiva ou Receptiva, em Razão e Cálculo Matemáticos, como na Linguagem Corporal e Social. Não tem como causa falta de interesse, de motivação, de esforço ou de vontade, como nada tem a ver com acuidade visual ou auditiva como causa primária. A Dislexia tem base neurológica, e que existe uma incidência expressiva de fator genético em suas causas
Disgrafia

Disgrafia é o transtorno da escrita, de origens funcionais, que surge nas crianças com adequado desenvolvimento emocional e afectivo, onde não existem problemas de lesão cerebral, alterações sensoriais ou história de ensino deficiente do grafismo da escrita. A criança disgráfica é vítima de transtornos que provém ora do plano motor, ora do plano perceptivo, ora do plano simbólico. A dificuldade de integração visual-motora dificulta a transmissão de informações visuais ao sistema motor. “A criança vê o que quer escrever, mas não consegue idealizar o plano motor”. Sua escrita é nitidamente diferente da escrita de uma criança que não tenha esse transtorno, o que não acarreta homogeneidade no interior do grupo dos disgráficos.
Disortografia

A Disortografia caracteriza-se por troca de fonemas na escrita, junção (aglutinação) ou separação indevidas das palavras, confusão de sílabas, omissões de letras e inversões. Além disso, dificuldades em perceber as sinalizações gráficas como parágrafos, acentuação e pontuação. Devido à essas dificuldades o indivíduo prepara textos reduzidos e apresenta desinteresse para a escrita. A Disortografia não compromete o traçado ou a grafia. Um sujeito é disortográfico quando comete um grande número de erros. Até a 2ª série é comum que as crianças façam confusões ortográficas porque a relação com sons e palavras impressas ainda não estão dominadas por completo.
Dispraxia

Dispraxia é uma disfuncão motora neurológica que impede o cérebro de desempenhar os movimentos corretamente. É a chamada "síndrome do desastrado". Seus sintomas são a falta de coordenação motora, falta de percepção e equilíbrio. A criança "dispráxica" tem uma falta de organização do movimento. As áreas que sofrem mais alterações são as do esquema corporal e a orientação temporo-espacial. Em alguns casos a linguagem não é afetada, a criança com dispraxia apresenta fracasso escolar, pois a escrita é a área mais comprometida.
Nenhum comentário:
Postar um comentário